

2D Ising model
Picture come from Wiki
目前關於 QUBO, 量子退火與 QAOA 演算法的教學文章仍在草稿階段圖中每一個箭頭都是一個 qubit,箭頭向上向下代表 qubit 處於的狀態($|0\rangle$ 或 $|1\rangle$),我們可以輕易地透過外加此場改變所有 qubits 的狀態,而量子退火便是透過這樣的原理尋找最優解。 一開始先設定後每個 qubit 的初始態,當下這系統的能量,我們記做 $H_i$,而我們欲求某個 QUBO 的問題記做 $H_p$,接著透過外加磁場慢慢地把系統能量從 $H_i$ 改變到 $H_p$,而外加磁場的改變會影響到每個 qubit 的狀態,這段過程每個 qubit 的變化會像下圖最下方的藍線一樣,從最左邊沿著藍線演化到右邊,之所以要「緩慢」變化是因為我們要求能量最小的組合,如果演化太快,系統可能會越躍遷到激發態,那就不是能量最低的組合,到了最右邊後,當下每個 qubit 的狀態就是能量最低的組合,在蛋白質模擬中,可能就對應每個氨基酸的位置。

退火示意圖
Picture come from D-Wave
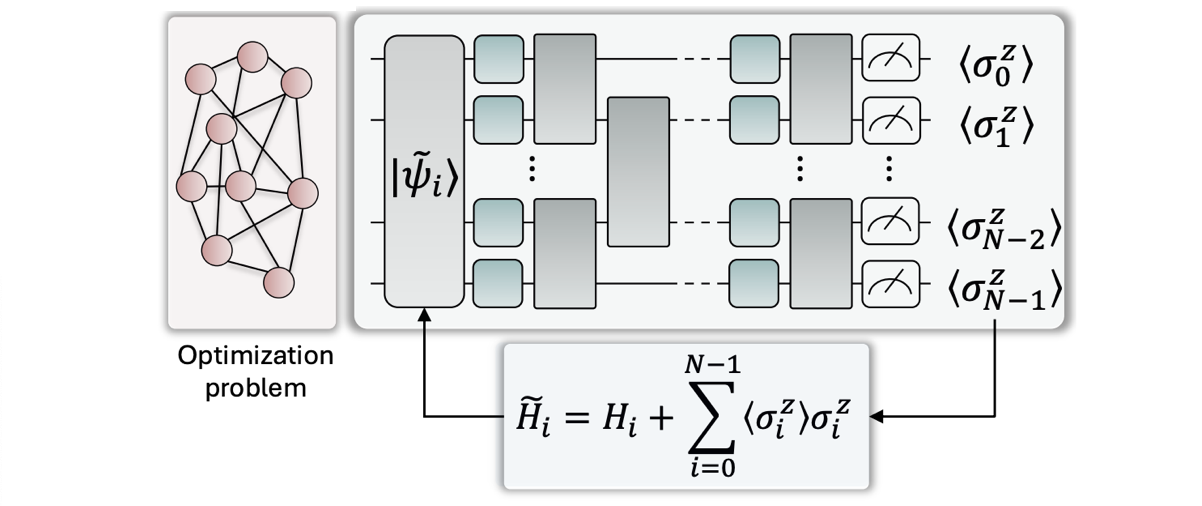
抵銷項的來由就是計算系統跳到激發態的機率,然後把它扣掉,實務上也是透過外加磁場實現(在量子退火裡)不過 IonQ 不是做量子退火,所以得把量子退火的過程分解成一個一個 quantum gate 來擬似量子退火,經過分解後,(1)式中的 $H_i$ 對應到下圖紫色方塊,[$R_y$](https://www.entangletech.tw/lesson/basic-algorithm-10#toc-7) gate,剩下的部分對應到下圖大括號中的 gate(包括綠色與灰色方塊)並重複 $n$ 遍,取決於你希望答案算得多精準。

BF-DCQO 演算法量子電路示意圖

BF-DCQO 演算法示意圖
上圖中敘述這種初始態更新方法的意思是,上一次測量 qubit 的結果,如果該 qubit 的量子態很接近 1,那再下一次計算時就讓該 qubit 的量子態設定得更接近 1(機率上更接近 1),中括號刮起來的部分,作者定義了兩種方法,詳情可以看參考文獻第二篇另外,如果對應出來的 gate 角度太小,對整體的計算影響不大,為了節省硬體資源可以將這 gate 省去,在論文中作者會設定一個閾值,小於這閾值的 gate 就刪掉,閾值越大代表刪掉的 gate 越多,視不同問題,作者會設定兩個閾值,閾值比較大的稱作 "hard",反之比較小的稱作 "soft"。 ## 蛋白質模擬 在簡單地介紹過以上兩個論文中提及的概念,現在我們把話題拉回蛋白質的模擬。首先,蛋白質的折疊本質上是一種搜尋能量最低的問題,這種問題通常需要在巨大的空間裡找到最佳或是接近最佳的解決方案,在蛋白質的摺疊中,尋找的就是在晶格上最低能量的空間(相對)位置。 蛋白質摺疊建模之優化問題通常涉及在二維或三維晶格上搜尋最低能量構象。這可以重構為量子基態問題。論文中採用的蛋白質建構方法為: 將氨基酸排列在四面體晶格(tetrahedral lattice)中,將蛋白質的第一個氨基酸放置於晶格中某個位置,接著就像貪吃蛇遊戲,下一個氨基酸就把它放在上、下、左、右四個方向之一的鄰近格子裡,方向用 2 個 qubit 來表示,因為 2 qubit 可以表達 4 種狀態($00、01、10、11$),這種編法方式稱作 dense encoding

蛋白質模擬的四面體晶格
Picture comes from doi: 10.1038/s41534-021-00368-4


實驗結果





